📝 Paper Summary
Protein Design
Generative Models for Biology
Drug Discovery
PepFlow is a multi-modal conditional flow matching model that co-designs peptide sequences and full-atom structures (including side-chains) to bind to specific protein targets.
Core Problem
Existing generative models for proteins often focus only on backbones or ignore the specific geometric constraints of peptide-protein binding, failing to model crucial side-chain interactions and full-atom details necessary for high-affinity binders.
Why it matters:
- Peptides are promising drug candidates due to high affinity and low toxicity, but the design space is too vast for traditional mutagenesis.
- Protein-peptide interactions rely heavily on side-chain dynamics, not just backbone positioning, making full-atom modeling essential.
- Current state-of-the-art methods like RFDiffusion primarily model backbones, often requiring separate steps for sequence design and side-chain packing which can lead to inconsistency.
Concrete Example:
When designing a binder, a backbone-only model might place a residue where its side-chain would clash with the target receptor because it doesn't explicitly model the side-chain angles (chi angles) during generation. PepFlow models these angles on a torus manifold to ensure geometric feasibility.
Key Novelty
Multi-Modal Riemannian Flow Matching for Full-Atom Peptides
- Decomposes a peptide residue into four modalities: backbone position (R3), orientation (SO(3)), side-chain torsion angles (Hypertorus), and residue type (Simplex).
- Constructs specific flow matching objectives for each manifold: Gaussian paths for positions, geodesic paths for rotations/torsions, and linear interpolation on logit space for discrete residue types.
- Jointly learns these flows conditioned on the target receptor structure, enabling simultaneous generation of sequence and full-atom structure.
Architecture
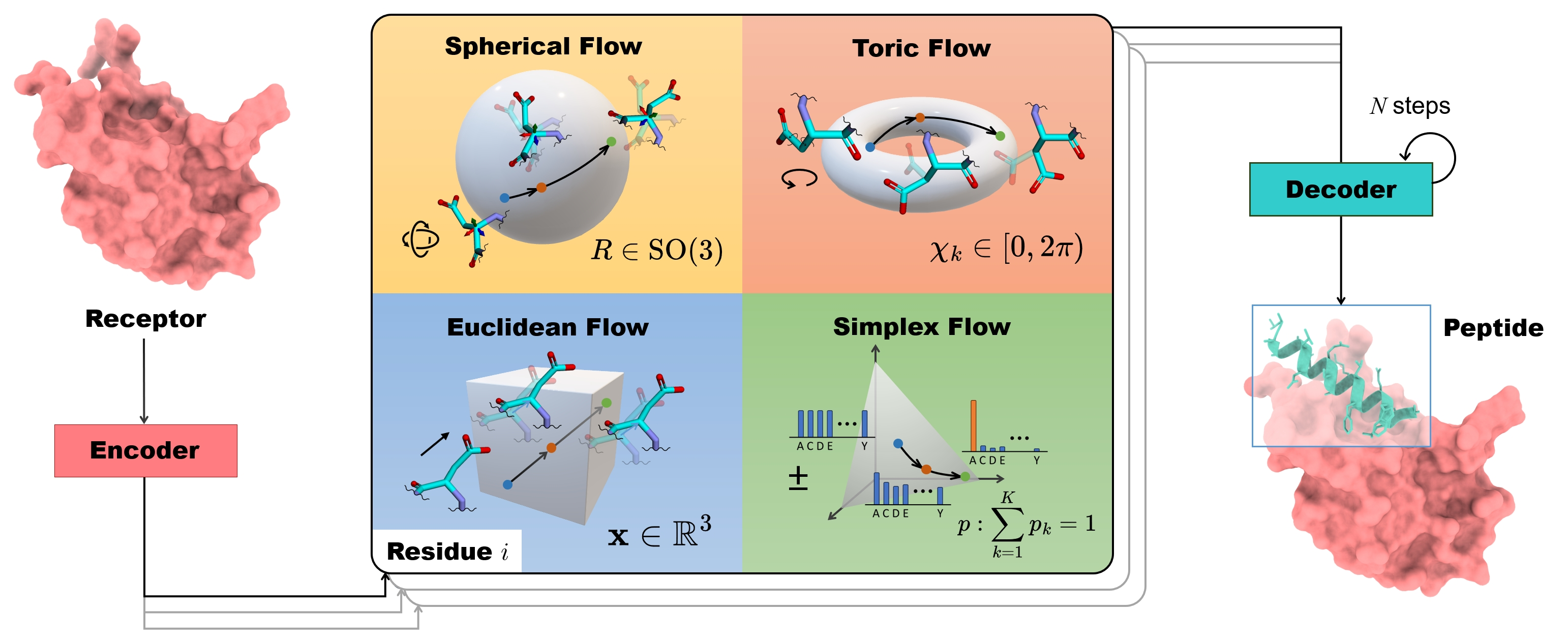
The overall framework of PepFlow. It illustrates the conditional generation process where a target protein is encoded, and the peptide is generated by transforming prior distributions (noise) on different manifolds (R3, SO(3), Torus, Simplex) into the data distribution using flow matching.
Evaluation Highlights
- Achieves lower (better) AAR (Amino Acid Recovery) perplexity than localized distributions, indicating generated sequences are plausible.
- Demonstrates high structural consistency with self-consistency scRMSD of 2.12 Å (lower is better), outperforming random baselines.
- Outperforms standard physics-based tools (like Rosetta FlexPepDock) in side-chain packing accuracy by directly modeling torsion angle distributions.
Breakthrough Assessment
7/10
Significant methodological advance in applying flow matching to complex, multi-modal biological manifolds. While experimental validation is wet-lab pending, the rigorous mathematical formulation for full-atom generation is a strong contribution.