📝 Paper Summary
Graph Neural Networks (GNNs)
Self-supervised Learning
Molecular Property Prediction
GraphFP pretrains GNNs by contrasting molecular graph node aggregates against chemically meaningful fragment graph embeddings, ensuring multiresolution structural understanding without privileged 3D data.
Core Problem
Existing molecular pretraining is either node-level (missing high-order structure) or graph-level (oversmoothing details), while current fragment-based methods use rigid rules or suboptimal embeddings.
Why it matters:
- Molecular datasets are small and data-hungry GNNs struggle to generalize without effective self-supervised pretraining on unlabeled data.
- Accurate property prediction (e.g., toxicity, permeability) requires capturing both local atomic interactions and global structural arrangements of functional groups.
- Many existing methods rely on expensive 3D coordinates or chemically invalid augmentations, limiting applicability.
Concrete Example:
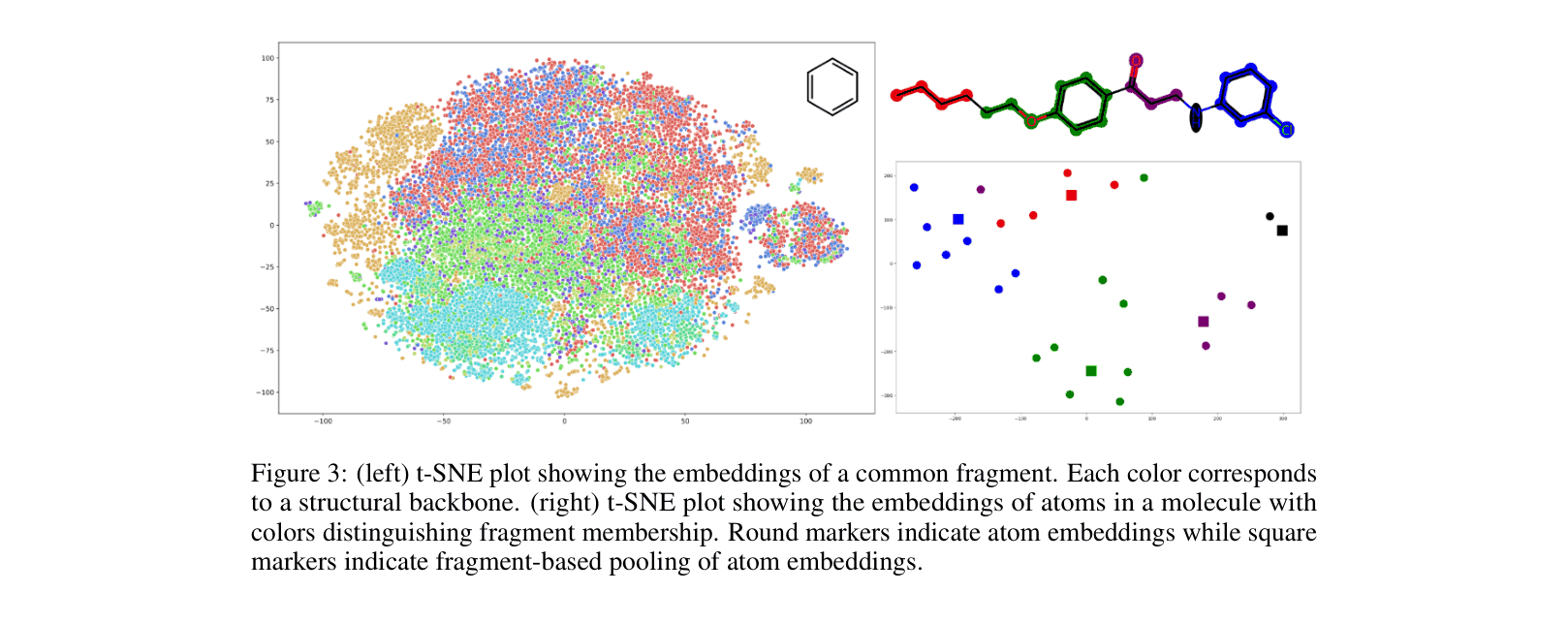
In predicting blood-brain barrier permeability (BBBP), standard GNNs fail to recognize how distant functional groups (like two -OH groups) interact spatially within a large molecule because they only aggregate local neighborhoods.
Key Novelty
Graph Fragment-based Pretraining (GraphFP)
- Constructs a separate 'fragment graph' where nodes are principal subgraphs (e.g., benzene rings) to explicitly model high-order connectivity.
- Contrasts the embedding of a fragment node against the aggregated embedding of its constituent atoms from the molecular graph, enforcing consistency across resolutions.
- Utilizes both the molecular encoder and the fragment encoder during downstream finetuning to combine local and global signals.
Architecture
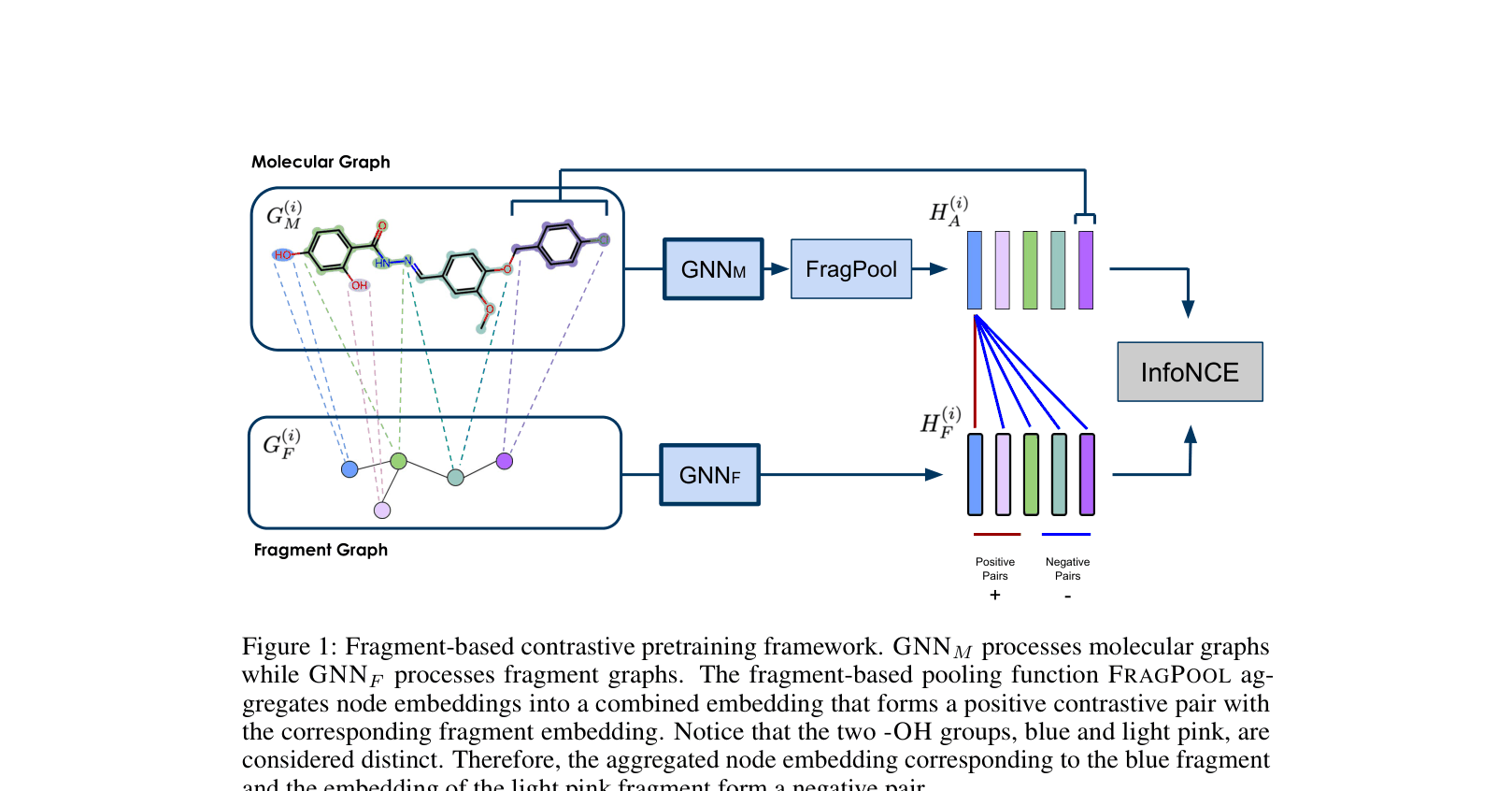
The contrastive pretraining framework. Two GNNs process the molecular graph and the fragment graph respectively.
Evaluation Highlights
- Achieves best performance on 5 out of 8 MoleculeNet benchmarks, outperforming contrastive baselines like GraphCL and JOAO.
- +14% improvement in Average Precision on the PEPTIDE-FUNC long-range benchmark compared to vanilla GIN (Graph Isomorphism Network).
- Reduces Mean Absolute Error by 11.5% on PEPTIDE-STRUCT compared to GIN, demonstrating superior capture of global structural arrangements.
Breakthrough Assessment
7/10
Strong empirical results on long-range tasks and standard benchmarks. The dual-graph approach is a logical evolution of motif-based learning, though reliance on heuristic fragmentation is a known technique.