📝 Paper Summary
Molecular Representation Learning
AI for Science
Foundation Models
Uni-Mol2 demonstrates that molecular representation learning follows scaling laws by training a 1.1-billion parameter model on 884 million 3D conformations, achieving significant gains on downstream tasks.
Core Problem
Existing molecular pretraining models are small-scale compared to NLP/CV models, and it is unknown if 'scaling laws' (performance improving with size/data) apply to molecular representation learning.
Why it matters:
- Traditional fingerprint methods fail to capture fine-grained structural features of large or complex molecules.
- Current pretraining explorations are limited to small datasets and architectures (e.g., GIN, SchNet), missing the potential benefits of large foundational models seen in other AI fields.
- Demonstrating scaling laws in this domain justifies the investment in massive datasets and compute for drug discovery and material science.
Concrete Example:
When predicting properties for complex molecules, small-scale models like GIN or fingerprints struggle with 3D spatial dependencies. Uni-Mol2 improves property prediction accuracy by 27% on QM9 by leveraging massive scale and 3D geometric pretraining.
Key Novelty
Billion-Scale Molecular Foundation Model (Uni-Mol2)
- Curates the largest 3D molecular dataset to date (884 million conformations) to fuel large-scale training.
- Systematically verifies scaling laws in molecular learning, showing power-law correlations between validation loss and model/dataset size.
- Scales the Two-Track Transformer architecture to 1.1 billion parameters, integrating atomic, graph, and geometric features.
Architecture
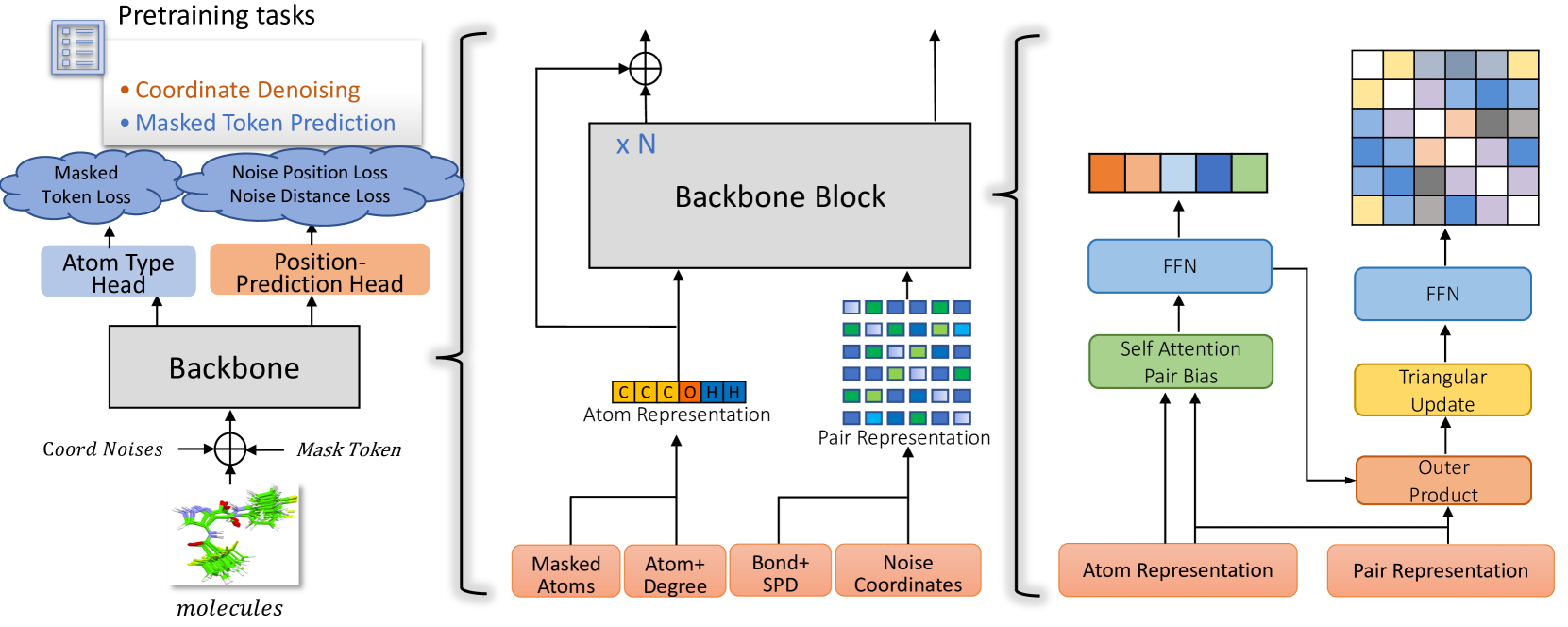
The Uni-Mol2 architecture, which is a two-track transformer processing atom and pair features in parallel.
Evaluation Highlights
- Achieves an average 27% improvement on the QM9 benchmark compared to existing methods using the 1.1B parameter model.
- Achieves an average 14% improvement on the COMPAS-1D dataset with the largest model.
- Demonstrates consistent power-law scaling behavior where validation loss decreases predictably as model size (84M to 1.1B) and data size increase.
Breakthrough Assessment
9/10
Represents a significant milestone as the first billion-scale molecular pretraining model, empirically establishing scaling laws in this domain and achieving substantial improvements on standard benchmarks.